El cobre es una sustancia esencial para el funcionamiento celular, sin embargo, su acumulación en los tejidos es extremadamente tóxica y puede producir daño celular irreversible (8). Existen muchas enfermedades relacionadas con la acumulación del cobre en el organismo y entre ellas destaca la Enfermedad de Wilson, un trastorno metabólico hereditario (5)(6) que lesiona en forma insidiosa y progresiva(9)(10) el hígado, los ganglios basales, las córneas y los riñones entre otros órganos (1)(5)(6)(7)(8)(9).
Fue publicada como una degeneración lenticular progresiva y enfermedad nerviosa familiar acompañada de cirrosis hepática(6) por Kinnier Wilson en 1912 (8)(9). Otros científicos como Westphal (1883), Strumpell (1898) y Gowers (1906) ya habían documentado casos con características clínicas similares bajo el nombre de pseudoesclerosis o como una corea tetanoide (6). Incluso los anillos corneales de color pardo, que algunos consideran como un signo patogmónico de la enfermedad, fueron descritos antes de la tesis doctoral de Wilson(6)(8). Los investigadores Kayser (1902) y Fleischer (1903) fueron quienes documentaron este signo clínico y lo asociaron a la pseudoesclerosis descrita por Westphal y Strumpell en años anteriores(6)(8).
Pero fue Kinnier Wilson el primer investigador en hacer referencia a la relación que existía entre la cirrosis y los trastornos neurológicos (6)(8), haciendo una descripción clínica y patológica mucho más completa que sus antecesores(7)(8). Su estudio estuvo basado en cuatro pacientes que sufrían de la misma enfermedad y ocho casos similares que había encontrado en la literatura existente hasta ese momento (3). Hizo referencia a la corta edad de inicio y a la afectación del sistema extrapiramidal(7). Sus conclusiones contenían la hipótesis que las anormalidades en el cerebro que causaban los movimientos involuntarios tenían que estar asociadas a una toxina generada en el hígado con cirrosis (7).
En 1913 Rumpell descubrió que el contenido de cobre en el hígado y en el encéfalo estaba aumentado, pero este hallazgo se ignora hasta que Mandelbrote (1948) lo asocia también a la excreción urinaria aumentada del cobre (6)(8). Estudios clínicos de Hall (1923) y Spielmeyer (1920), basados en los cortes de hígado y encéfalo de Westphal y Strumpell, establecen que la pseudoesclerosis es en realidad la enfermedad descrita por Wilson en 1912(6). En este momento es cuando se hace la relación entre los Anillos de Kayser-Fleischer y la Enfermedad de Wilson. Luego, en 1953, Scheinberg y Gitlin descubren que la ceruloplasmina está muy reducida en la enfermedad (6), lo cual constituye un hallazgo que hoy en día permite sospechar la existencia de la enfermedad en ciertos pacientes.
La Enfermedad de Wilson también es conocida también como degeneración hepatolenticular (1)(9)(10) y es invariablemente fatal si no es tratada a tiempo (4)(7)(10). Actualmente se calcula una incidencia de 1:30,000 (1)(4)(5)(8)(10) siendo mayor en áreas con alto grado de consanguinidad(1)(10). Japón tiene la proporción más alta a nivel mundial, pues alcanza un valor de 1:10,000 (4). Cuando ambos padres son portadores de los genes afectados, el riesgo de padecer la enfermedad se calcula en 1:200 (6)(10).
La prevalencia en Costa Rica estaba calculada en 1:25,000 habitantes en 1989 (10), pero según estudios recientes actualmente se estima en 1:15,000 (4). Según la Asociación Costarricense de Pacientes con Enfermedad de Wilson, la mayoría de los casos están ubicados en Puriscal, Acosta, Aserrí y Santa María de Dota.
Genética
La Enfermedad de Wilson es un trastorno autosómico recesivo(1)(4)(5)(6)(10) que provoca una mutación del gen ATP7B (1)(4)(7)(8), localizado en el cromosoma13q14 (4)(5)(6)(9). Se han detectado más de 300 mutaciones (6)(8)(9) y hasta ahora no se ha demostrado si la cantidad de variaciones existentes tiene relación con la diversidad de formas en que la enfermedad se manifiesta (8).

Cromosoma 13
Tomado de http://www.herenow4u.net/
Tomado de http://www.herenow4u.net/
Al igual que otras enfermedades autosómicas recesivas, la expresión del defecto tiende a ser más uniforme que en las autosómicas dominantes (5). La penetrancia completa es frecuente y el inicio de la enfermedad es temprano (5). En los heterocigotos no muestran el fenotipo de la enfermedad pues sintetizan enzimas normales y defectuosas, siendo las enzimas normales las utilizadas por el organismo (5).
La mutación H1069Q es la más frecuente en Estados Unidos y Europa (8) y al menos un estudio lo relaciona con las manifestaciones tardías (6). En los estudios realizados por Duarte en el 2009 (1) y por Schoskinski en 1989 (10) no se mencionan si esta u otra variación de la enfermedad predomina en Costa Rica, sin embargo en la investigación de Jiménez del 2008 se menciona la presencia de una mutación que es común en Sardinia, Italia(4) y que puede ser correlacionada con inmigrantes de ese país según los registros históricos.
Se calcula que un 1% de la población es portadora de alguna de las mutaciones del gen ATP7B (8).
Fisiopatología
El cobre es esencial para el funcionamiento de una cantidad de enzimas (1)(2)(8). El promedio normal de absorción es de 1,5 a 5 mg (1)(5) y se realiza principalmente en el estómago y duodeno (1)(7) en donde se une a la albúmina para ser transportado al hígado (5), pues las células hepáticas tienen gran afinidad por esta proteína unida al cobre (1).
Una vez que el cobre ingresa al hígado, el metal se incorpora con una globulina para formar la ceruloplasmina (1), un proceso que depende de una ATPasa tipo P de iones metálicos llamada ATP7B (1). Esta proteína está codificada a partir del gen que lleva el mismo el nombre (1)(7)(8) y como transportador está localizado en la región de Golgi de los hepatocitos (5)(8). Tanto la proteína ATP7B como la ceruloplasmina son considerados elementos claves en la movilización de cobre en el organismo (8).
La ceruloplasmina se secreta al plasma sanguíneo transportando aproximadamente el 90 % a 95% del cobre total (5). Esto significa que las cantidades libres del metal en condiciones normales deben ser sumamente bajas. La parte final del proceso se lleva a cabo en el hígado, en donde la ceruloplasmina es degradada por lisosomas y el cobre es secretado en la bilis (5).
En la Enfermedad de Wilson, la absorción y transporte del cobre al hígado son normales (5)(10) sin embargo el cobre absorbido no consigue entrar en la circulación en forma de ceruloplasmina pues la proteína ATP7B mutada no realiza su función en forma adecuada. Esto provoca que la secreción biliar de cobre desde los lisosomas hepatocelulares sea marcadamente disminuida (5)(10).
En resumen, la función defectuosa del ATP7B conlleva a un fallo en la secreción de cobre en la bilis (8) que es la principal vía de eliminación del cobre corporal (5). La acumulación excesiva de cobre en los diferentes tejidos es la responsable de la disfunción hepática, la alteración renal, los trastornos neurológicos y de la formación de los anillos de Kayser-Fleischer (1)(5)(10). También se han reportado cambios a nivel óseo (2)(10), articular(5), en las glándulas paratiroides(5) y de la piel (2). En forma menos frecuente se observan trastornos psicológicos, esplenomegalia aislada y crisis hemolíticas causadas por la lesión de los eritrocitos (1)(7)(10). En el caso particular del tejido cerebral, el daño tóxico afecta principalmente a los ganglios basales (putamen), los cuales muestran atrofia y cavitación (5).
Existen un 5% de casos donde una proteína anormal con alta afinidad por el cobre impide la unión del catión con la apoceruloplasmina (10) y algunos autores asocian la Enfermedad de Wilson con un déficit de dopamina a nivel cerebral (9).

Acumulación de cobre en el tejido hepático de un paciente con Enfermedad de Wilson
Tomado de http://www.medscape.com/
Tomado de http://www.medscape.com/
Clínica
Los síntomas iniciales de la enfermedad suelen ser hepáticos, neurológicos o psiquiátricos (4)(6). El grado de afectación de cada uno de ellos varía, sin embargo algunos autores coinciden en que las lesiones hepáticas y neurológicas son predominantes (1)(6). Rara vez son detectados antes de los 6 años (5)(7)(8), pues antes de esa edad el cobre libre no ha producido cambios patológicos significativos (5).
Las manifestaciones hepáticas tienden a ocurrir primero (1)(7)(8) en un 40 a 50% de los casos (8), con la edad de inicio promedio para los pacientes con alteraciones de este tipo de problemas es de 11 años (8). Su aparición luego de la cuarta década es poco frecuente (8).
Entre las principales manifestaciones hepáticas se encuentran:
- Hepatitis aguda o crónica (1)(4)(5)(7)(8)(10)
- Hepatomegalia por elevación de las enzimas hepáticas (1)(8)
- Hipertensión portal con hiperesplenismo (1)
- Diátesis hemorrágica (1)
- Ictericia (1)(7)
Entre las principales manifestaciones neurológicas se encuentran:
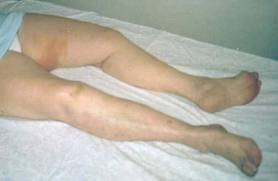
- Temblor/Tremor (1)(4)(6)(7)(8)(9)
- Distonía (1)(7)(8)(9)
- Disartría (1)(4)(6)(7)(8)(9)
- Alteración de la marcha(9)
- Risa sardónica (7)(9)
- Rigidez (6)(7)(9)
- Disfagia (1)(6)(9)
- Problemas de coordinación (1)(7)(6)
- Sialorrea (7)(8)
- Ataxia (7)
- Corea y atetosis (8)(9).
Las manifestaciones psiquiátricas se presentan en forma aislada en un 20% de los casos, lo cual hace muy difícil su diagnóstico (4)(6). Los cambios de personalidad son de inicio muy sutil y tienden a aparecer en la segunda década de vida (1)(5)(7).
Entre las principales manifestaciones psiquiátricas se encuentran:
- Depresión (6)(7)(8)(9)
- Irritabilidad(1)(9)
- Agresividad(9)
- Esquizofrenia (6)(9)
- Inhibición Sexual (7)(8)
- Psicosis (9)
- Deterioro en el rendimiento escolar y laboral (7)(8)
- Ansiedad y angustia (7)
- Psiconeurosis (7)
Se menciona la necesidad de tener un cuidado especial al diagnosticar psicosis. Existen casos descritos en la literatura médica donde se determina en forma errónea la esquizofrenia es la enfermedad de fondo y se cree que los signos extrapiramidales son consecuencia del uso de antipsicóticos (9).

Anillo de Kayser-Fleicher
Tomado de http://ocularis.es/
Se mencionan también otras manifestaciones sin que haya realmente un consenso respecto a las estadísticas de aparición, por ejemplo:
- Anemia Hemólitica (1)(4)(8)(10)
- Trombocitopenia (4)
- Disfunción de los túbulos renales (1)(4)(7)(8)
- Esplenomegalia (4)(8)
- Hiperpigmentación (8)
- Irregularidad menstrual (6)(7)(8)
- Anorexia (1)
- Fatiga (1)(7)
- Problemas osteoarticulares (7)(8)
- Cardiopatías y arritmias (7)
Diagnóstico
La sospecha de la Enfermedad de Wilson debe ser apoyada por un diagnóstico bioquímico basado principalmente en (5)(9)(10):
- Disminución de ceruloplasmina sérica
- Aumento hepático de cobre
- Incremento en la secreción urinaria de cobre
Dado que la distribución de cobre en el hígado es irregular y que podrían existir problemas de coagulación, la biopsia hepática podría no indicar un contenido anormal de ese metal en los tejidos (6)(10). Sin embargo, encontrar cobre en la prueba se considera un método relativamente confiable para detectar la enfermedad (8).
La cantidad de mutaciones documentadas hacen que las pruebas genéticas sean imprácticas (8), mientras que reportes recientes han demostrado la presencia de anormalidades a nivel de los ganglios basales en un 100% de los pacientes con afectación neurológica. Es por ello que se favorece el uso de la resonancia magnética (MRI) para apoyar el diagnóstico especialmente cuando hay manifestaciones neurológicas tempranas (8).
A nivel pediátrico, la triada de ceruloplasmina disminuida, anillos de Kayser-Fleischer y hepatitis se presenta en el 30 a 50% de los casos (4).

MRI de un paciente con afectación neurológica
Tomado de http://ocularis.es/
En Costa Rica se realizó un estudio entre 1975 y 1982 con 61 pacientes con el objetivo de determinar los hallazgos de laboratorio más importantes para el diagnóstico (10). La mayoría se manifestaron clínicamente antes de la tercera década(10). Todos los pacientes mostraron un nivel de ceruloplasmina inferior al intervalo de referencia (55 a 150 U/L) y 43 de ellos mostraron ausencia total de la proteína (10). El cobre sérico se mostraba disminuido en la mayoría de los pacientes mientras que el cobre urinario se encontraba aumentado en la mayoría de los casos (10) A 21 pacientes se les practicó una biopsia o necropsia hepática, mostrando todos ellos valores entre 20 y 147 mg/100g lo cual representa valores altos respecto al intervalo de referencia de 0-6 mg/100g (10).
Entre 1992 y el 2006, se realizó una revisión retrospectiva de 35 expedientes de niños diagnosticados en el Hospital Nacional de Niños de Costa Rica y se compilaron las características demográficas y su presentación clínica (4). Un 70% correspondía a varones y la edad promedio de presentación de los síntomas de 10 años de edad (4). La presentación hepática se detectó en 69% de los pacientes, 11% presentaron afectaciones hematológicas y solamente uno de los niños presentó problemas neurológicos (4). La historia clínica de este paciente reveló una regresión de la motora gruesa, un escaso desarrollo escolar y un progreso rápido hacia la espasticidad y disartria (4).
El diagnóstico diferencial de la Enfermedad de Wilson se realiza contra cualquier enfermedad que provoque un aumento en el cobre sérico, por ejemplo la colestasis obstructiva crónica (1)(5)(10), cirrosis biliar primaria (10), atresia biliar (10), Enfermedad de Menkes (2)(6)(10) y sobre todo las hepatitis agudas o crónicas de origen viral (1)(5)(10). También es necesario descartar la hepatitis autoinmune (8), la distonía de torsión idiopática (7), el parkinsonismo juvenil (7) y la coreoacantocitosis (7).
Tratamiento, rehabilitación y pronóstico
El diagnóstico y tratamiento temprano es decisivo para evitar el daño permanente al hígado y al cerebro (6)(7). Los pacientes con síntomas neuropsiquiátricos tienen un diagnóstico más tardío y por lo tanto su pronóstico es más desfavorable que aquellos que tienen sólo presentación hepática (6).
Los dos enfoques para el tratamiento son los agentes quelantes como Penicilamina y la Treintina (1)(4)(6)(8) o la prevención de la absorción de cobre mediante el uso de altas dosis de zinc (1)(4)(6)(8). Los primeros ayudan a eliminar el excedente de cobre por la vía urinaria sin embargo el cobre acumulado en el hígado no se remueve por completo (6). También se sugiere disminuir la ingesta de alimentos ricos en cobre como el chocolate, nueces y mariscos (1).
El transplante hepático es la opción final en caso de insuficiencia fulminante de ese órgano o con manifestaciones neurológicas resistentes al tratamiento (4)(6).
La rehabilitación de los pacientes con afecciones neurológicas busca que estos logren desenvolverse lo mejor posible en su medio social. Para ello su atención debe darse de forma integral, que incluya tanto especialistas médicos, terapeutas físicos, ocupacionales y psicólogos (7). La práctica y la repetición de los ejercicios en patrones de movimiento normales son los dos principios en los que debe basarse la rehabilitación de trastornos del sistema nervioso. El paciente deber ser estimulado en coordinación y control para desempeñar una marcha normal (7).
Conclusiones
La Enfermedad de Wilson, al igual que muchos trastornos metabólicos, requiere que los especialistas en salud sean sumamente cuidados en el diagnóstico, tratamiento y rehabilitación de los pacientes. Las manifestaciones hepáticas, neurológicas y psiquiátricas pueden ser fácilmente confundidas con otras patologías consideradas más comunes en ciertas áreas geográficas. Aunque existen signos muy característicos de la enfermedad, la cantidad de variaciones existentes hace obligatorio el análisis a profundidad de la historia clínica, signos y síntomas y por supuesto los hallazgos de laboratorio.
En Costa Rica, la incidencia de la enfermedad es mayor que en otras partes del mundo. Esto significa que todo profesional está en la obligación de sospechar esta enfermedad cuando existan trastornos hepáticos a edades tempranas especialmente si no hay antecedentes de alcoholismo. También es sumamente importante realizar los análisis bioquímicos sugeridos cuando existen afectaciones neurológicas o psiquiátricas sin causa aparente.
Principalmente en niños y adultos jóvenes, la probabilidad de mantener la enfermedad controlada y llevar una vida normal es bastante alta si se diagnostica el trastorno en sus primeras etapas. Estudios disponibles actualmente señalan la eficacia de los tratamientos farmacológicos y las ventajas de la rehabilitación física, que combinada con un apoyo multidisciplinario pueden mejorar considerablemente la calidad de vida de las personas que padecen la enfermedad.
Proveer información correcta y confiable a los pacientes y sus familiares es de vital importancia. Esto obliga a todo profesional a actualizarse constantemente y sobre todo buscar datos recientes sobre incidencia y tratamientos disponibles que podrían ayudar a los pacientes a mejorar su calidad de vida aun cuando su enfermedad se considere incurable.
Referencias Bibliográficas
(1) Duarte, Tatiana. 2009. Enfermedad de Wilson. Revista Médica de Costa Rica y Centroamérica. LXVII (590) 373-375 2009. San José, Costa Rica.
(2) González, J. Cocho, J.A. 2002. Metodología Recomendada para la medición del contenido de cobre en especímenes biológicos. Sociedad Española de Bioquímica Clínica y Patología Molecular. 2002; 21 (2) 62-66. Barcelona, España.
(3) Hoogenraad, T.U. 2001. Pionners in Neurology: S.A. Kinnier Wilson. Journal of Neurology. Volume 248. Number 1. 71-72. VD Doorn. Netherlands.
(4) Jiménez, Gabriela. Cambronero, Victor. Morales, Carlos. Mora, Alfredo. Guzmán, Celina. Jiménez, Carolina. 2008. Enfermedad de Wilson: Experiencia Pediátrica en Costa Rica. Gastroenterolgía y Hepatología. 2009;32:274-8. - vol.32 núm 04. San José, Costa Rica.
(5) Kumar, Vinay. Abbas, Abul. Fausto, Nelson. Mitchell, Richard. 2008. Robbins Patología Humana. Octava Edición. Capítulo 7: Enfermedades Genéticas y Pediátricas. Editorial Saunders. Barcelona, España.
(6) López Hernández, Marco A. Serrano, Rufino. Marlene. 2007. Enfermedad de Wilson: reporte de un caso y revisión de la literatura. Revista Medicina Interna de México. Volumen 23, Número 5. México.
(7) Madrigal Salas, Andrea. Zumbado Delgado, Carolina. 2009. Propuesta de Abordaje Fisioterapéutico para Pacientes con la Enfermedad de Wilson. Universidad Santa Paula. San José, Costa Rica.
(8) Pfeiffer, Ronald. 2007. Wilson’s Disease. Theme Medical Publishers. Aprill 27(2):123-132. New York, USA.
(9) Restrepo B, Diana. Calle B, Jorge J. 2007. Aspectos Neuropsiquiátricos de la Enfermedad de Wilson y la Esclerosis Múltiple. Revista Colombiana de Psiquiatría. Vol XXXVI. Suplemento 1. Bogotá, Colombia.
(10) Schoskinski, Kari. Vargas, Marianella. Esquivel, Alba Luz. Grant, Sonia. Artavia, Alfredo. Chavarría, Miguel. 1989. Hallazgos de Laboratorio en 61 Casos de Enfermedad de Wilson en Costa Rica. Revista Costarricense de Ciencias Medicas. Volumen 10, número 3. San José, Costa Rica.
(11) Tortora, Gerald. Derrickson, Bryan. 2006. Principios de Anatomía y Fisiología. 11ª. Edición. Editorial Médica Panamericana. México DF. México. Cap 6.






















{kind=link}
{kind=link}
{kind=link}
{kind=link}